
# 83 - Interrupción del arco aórtico fetal
# 83 - Interrupción del arco aórtico fetal
Fecha:
Septiembre 2022
Autores:
Magdalena Basso
Se presenta a la consulta una paciente secundigesta de 30 años de edad cursando embarazo de 27.4 semanas. Es derivada del interior del país por hallazgos sugestivos de patología cardíaca en el estudio morfológico del segundo trimestre. No presenta antecedentes de relevancia. Adjunta un screening de aneuploidías del primer trimestre con resultado bajo riesgo. Durante la evaluación ultrasonográfica cardíaca, se observaron los siguientes hallazgos: Figuras 1-4. Elija la opción correcta: a. Se observa una comunicación interventricular (CIV) de salida, sin otros hallazgos relevantes. b. Los hallazgos son compatibles con interrupción del arco aórtico de tipo B. c. Los hallazgos son compatibles con coartación aórtica (dominancia de cavidades derechas y alteración en el corte de 3 vasos). d. Los hallazgos son compatibles con tetralogía de Fallot con atresia pulmonar.
Respuesta correcta: B. Los hallazgos son compatibles con interrupción del arco aórtico de tipo B.
La interrupción del arco aórtico (IAA) es una malformación congénita poco frecuente, que se caracteriza por la total discontinuidad entre la porción ascendente y descendente de la arteria aorta. Su incidencia aproximada es de 2-3 por cada 1.000.000 nacidos vivos, que supone aproximadamente el 0,2 a 1% de todos los niños nacidos con cardiopatías congénitas (CC). Se enmarca dentro de las cardiopatías obstructivas del arco aórtico, junto a la coartación de aorta, con una frecuencia en el diagnóstico prenatal de aproximadamente el 50%. Esta cardiopatía se encuentra frecuentemente asociada a otras anomalías congénitas, tanto cardíacas como extracardíacas. Presenta también una fuerte asociación con la microdeleción 22q11.2.
La interrupción del arco aórtico es invariablemente preductal asegurando, de esta manera, la perfusión sistémica de la parte inferior del cuerpo fetal. Se clasifica en tres variantes según la relación entre el sitio de interrupción y los vasos que se originan en el arco aórtico:
Tipo A: La interrupción del arco aórtico es distal al origen de la arteria subclavia izquierda. Es responsable de un 30% a 40% de los casos. Su localización en muchos casos dificulta el diagnóstico diferencial con la coartación de aorta severa y ambas patologías tienen características hemodinámicas similares.
Tipo B: Es la variante más frecuente y representa un 50% - 60% de los casos. Está caracterizada por la interrupción del arco entre el origen de la arteria subclavia izquierda y la carótida común izquierda. Se asocia casi invariablemente con una comunicación interventricular (CIV) de tipo mal alineada con desplazamiento posterior del tabique infundibular. La válvula aórtica suele ser bicomisural y puede tener un grado variable de hipoplasia. Presenta una fuerte asociación con alteraciones en la alineación entre el tabique interventricular infundibular y el muscular, lo que genera estrechamiento en el tracto de salida del ventrículo izquierdo y la categoriza dentro de las cardiopatías conotruncales. Esta variante particularmente, en aproximadamente la mitad de los casos, se asocia a la microdeleción 22q11.2.
Tipo C: En estos casos, la interrupción del arco aórtico se encuentra entre el origen del tronco braquiocefálico y la carótida izquierda. Supone menos del 5% de los casos.
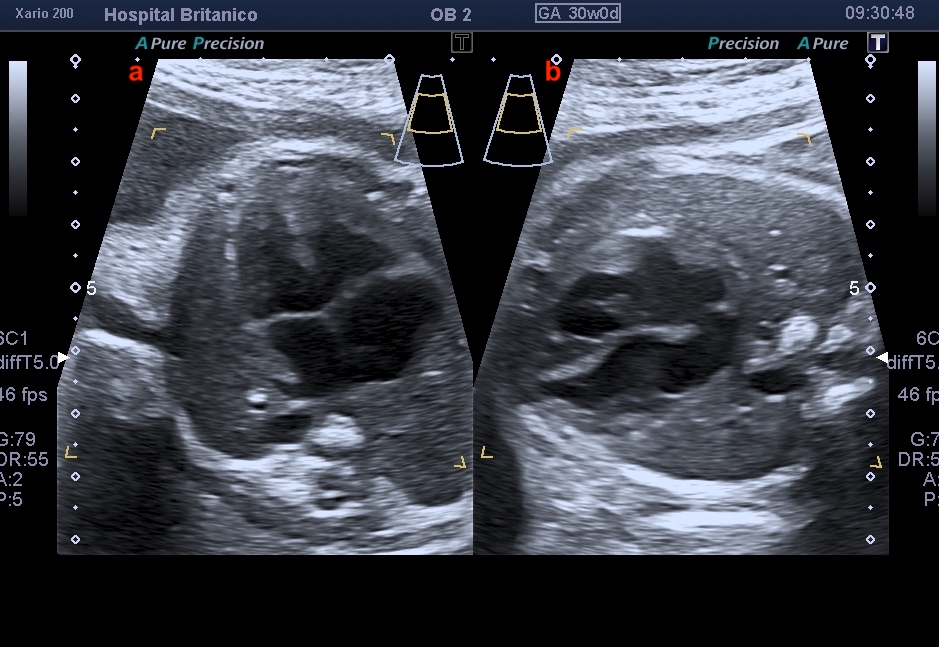
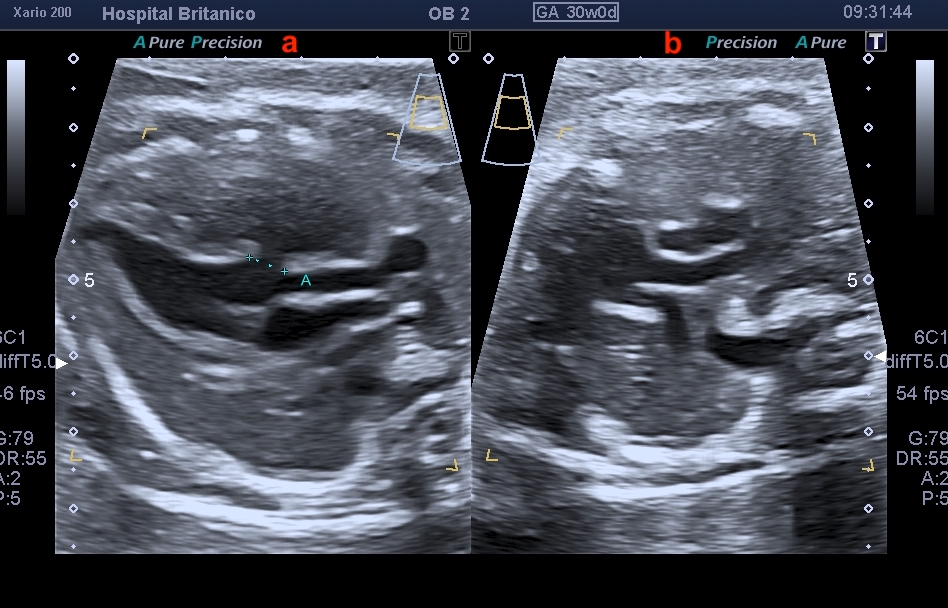
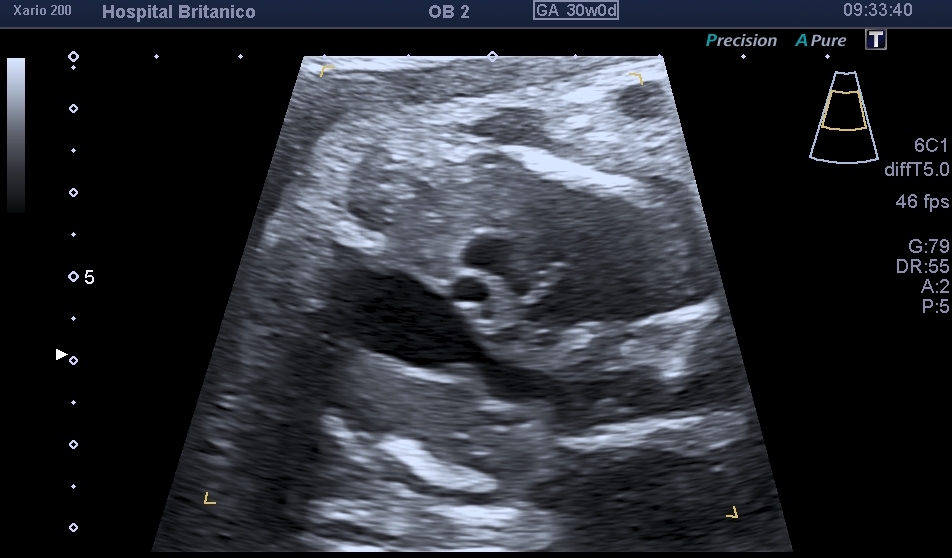
El diagnóstico prenatal se basa en la sospecha de esta cardiopatía mediante la visualización de signos ecográficos indirectos y su confirmación mediante signos directos. Los signos indirectos incluyen la asimetría de cavidades ventriculares en el plano de cuatro cámaras, a expensas de las izquierdas de menor tamaño, similar a lo observado en la coartación de aorta. Esta asimetría se observa también en el tracto de salida del ventrículo izquierdo, en donde se ve menor calibre de la aorta ascendente. Sin embargo, dado que el tipo B es el más frecuente, y en la mayoría de los casos coexiste con una CIV subaórtica, es habitual encontrarse con un plano de 4 cámaras normal (Fig. 1). La CIV, salvo que sea de gran tamaño, no se suele evidenciar en el corte de 4 cámaras, y consecuentemente, solo se evidencia la asimetría a nivel de los grandes vasos (Fig. 2). En el corte de los 3 vasos, es evidente la disminución del calibre de la aorta ascendente que es menor al calibre de la arteria pulmonar, pero más llamativamente, menor al calibre de la vena cava superior, observada en este mismo plano (Fig. 3). En el corte de 3 vasos y tráquea, no es posible evidenciar la continuidad del arco aórtico. Es frecuente observar una disminución del tamaño del timo, y aún su ausencia, dado que esta cardiopatía se encuentra muy asociada a la microdeleción 22q11.2.
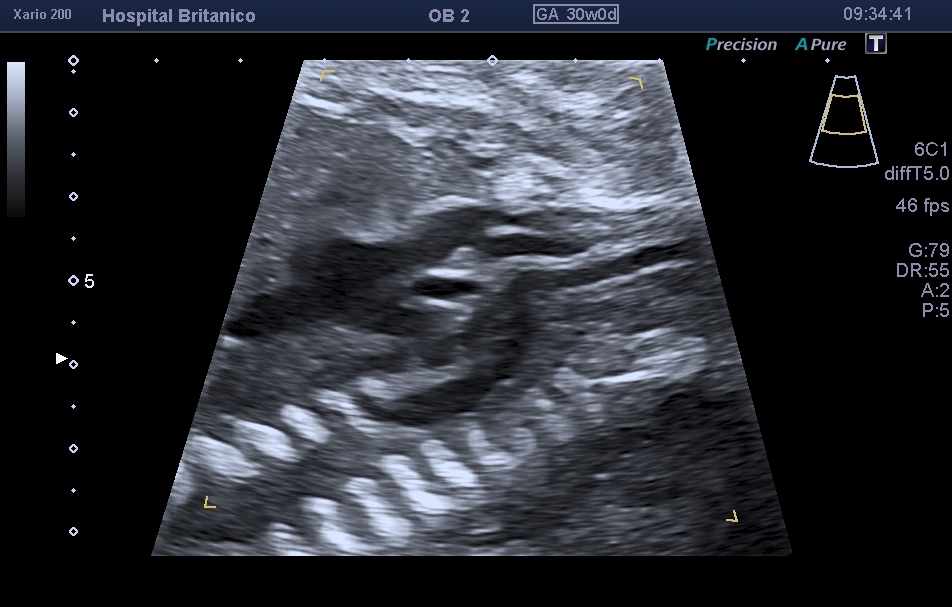
Los signos directos incluyen la visualización del arco aórtico en el corte sagital (Fig. 4). En el tipo B, se observa el arco aórtico rectificado con dos ramificaciones vasculares: el tronco braquiocefálico y la carótida común izquierda. En el tipo A el arco se interrumpe distal a la arteria subclavia izquierda, y se puede observar una discreta curvatura del arco con sus tres troncos supraaórticos. Este tipo es muy similar a la coartación de aorta, cardiopatía mucho más frecuente que la IAA tipo A.
El Doppler color es una herramienta útil para confirmar los hallazgos de la ecografía bidimensional.
El diagnóstico diferencial de esta cardiopatía es un desafío, en particular la diferenciación entre cada tipo de IAA. La presencia de la CIV subaórtica, es característica del tipo B y la asimetría de cavidades ventriculares puede orientar más para los tipos A y C, y el tipo A con la coartación de aorta.
Entre las cardiopatías congénitas asociadas se incluyen al arco aórtico derecho, la arteria subclavia derecha aberrante, transposición de grandes arterias y la ventana aorto-pulmonar.
Las malformaciones extracardíacas incluyen alteraciones del sistema nervioso central, urinario, esquelético y gastrointestinal. La IAA tipo B tiene una gran asociación con la microdeleción 22q11.2, y por lo tanto se deben buscar las relacionadas con ella, como la visualización de una hipoplasia tímica, micrognatia, etc. En todos los casos se debe realizar una evaluación anatómica detallada de todo el feto.
El manejo prenatal incluye una valoración ecocardiográfica detallada y el seguimiento de esta CC para valoración del crecimiento de las estructuras cardíacas, dado que se trata de una cardiopatía evolutiva en vida prenatal. El asesoramiento genético es importante para confirmar o descartar su asociación con la microdeleción 22q11.2.
Considerando que la IAA se enmarca dentro de las cardiopatías congénitas ductus dependientes, el diagnóstico prenatal es clave para el correcto manejo postnatal. Si bien el momento y modo del nacimiento no deben ser modificados, se debe asegurar el nacimiento en un centro de alta complejidad, con la administración de Prostaglandina E1 para mantener la permeabilidad ductal, y la posibilidad de realizar de manera precoz la reparación quirúrgica de la cardiopatía.
Lecturas Recomendadas:
1. Hanneman K, Newman B, Chan F. Congenital Variants and Anomalies of the Aortic Arch. Radiographics. 2017;37: 32–51.
2. Friedman K. Preoperative Physiology, Imaging, and Management of Interrupted Aortic Arch. Semin Cardiothorac Vasc Anesth. 2018;22: 265–269.
3. LaPar DJ, Baird CW. Surgical Considerations in Interrupted Aortic Arch. Semin Cardiothorac Vasc Anesth. 2018;22: 278–284.
4. Escribano D, Herraiz I, Galindo A. Defectos del corazón izquierdo. In: Galindo A, Gratacós E, Martinez JM, editors. Cardiología Fetal. MARBÁN; 2015. pp. 272–341.
5. Abuhamad A CR. Ecocardiografía fetal. Abuhamad A CR, editor. Ediciones Journal; 2019.






")
")