
# 75 - Cardiopatía conotruncal
# 75 - Cardiopatía conotruncal
Fecha:
Junio 2020
Autores:
Dra. Yamila Marques
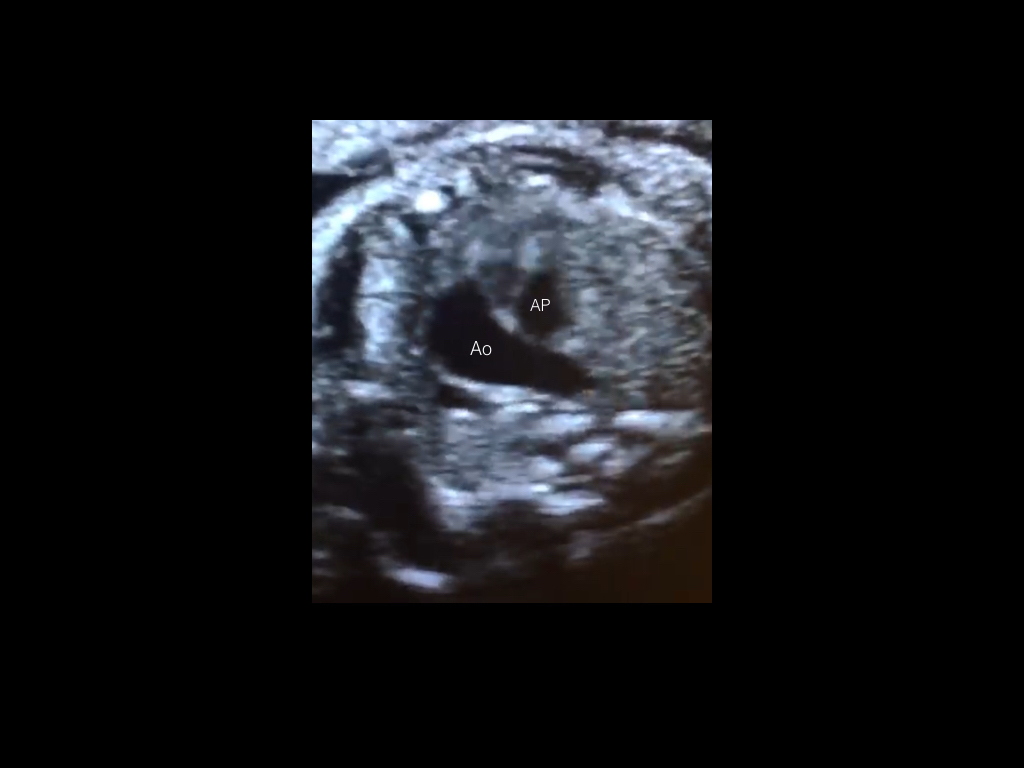
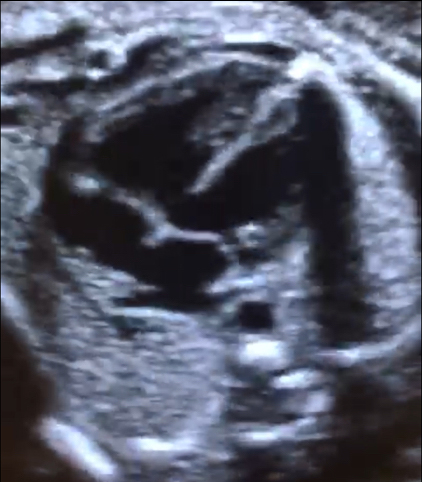
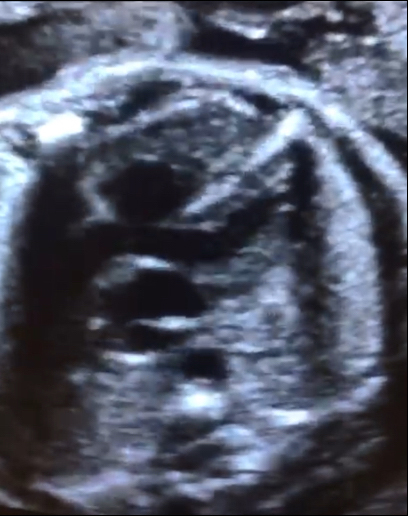
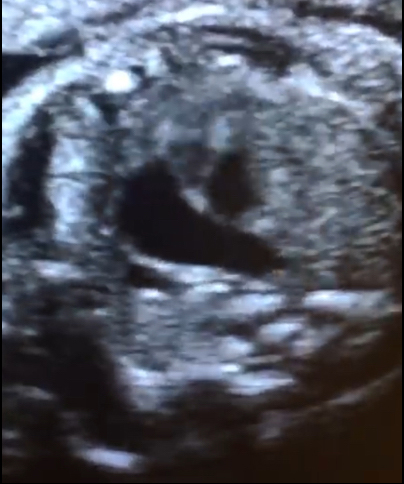
Se presenta a la consulta una paciente de 34 años de edad, sin antecedentes de relevancia, para realizar estudio morfológico fetal en semana 23. Se adjunta el estudio de screening de aneuploidias, de la semana 11-14, que informa bajo riesgo. Durante la evaluación del corazón fetal se observan los siquientes hallazgos: Figura 1: Vista apical del corte de cuatro cámaras cardíacas. Figura 2: Vista apical del corte de cinco cámaras cardíacas. Figura 3: Vista del corte de tres vasos. Elija la opción correcta: a) La anatomía del corazón fetal es de apariencia normal para la edad gestacional. b) A nivel del corte de cuatro cámaras se observa ausencia del septum primum y asimetría de cavidades. c) Se debe descartar la presencia de cardiopatía conotruncal.
.jpeg)
.jpeg)
Respuesta correcta: C. Se debe descartar la presencia de una cardiopatía conotruncal.
Las cardiopatías congénitas (CC) son las malformaciones congénitas severas más frecuentes. Sin embargo, la mitad de los casos de CC son leves y fácilmente corregibles quirúrgicamente, siendo el resto de los casos responsables de la mitad de las muertes por anomalías congénitas en la niñez. Afectan aproximadamente al 0.8 - 1% de los recién nacidos.
Las imágenes del caso clínico presentado corresponden a un feto con sospecha diagnóstica de tetralogía de Fallot.
Las cardiopatías conotruncales son un grupo heterogéneo de malformaciones que comprometen la región conotroncal y parte de los ventrículos, presentando una alta tasa de detección prenatal siendo siempre enfermedades ductus dependientes. Se originan debido a una alteración en la formación y tabicación de los tractos de salida y de los grandes vasos del corazón.
Estas anomalías representan entre el 20 al 30% % de todas las CC que se detectan en el período prenatal. La tetralogía de Fallot y la transposición de grandes vasos son las dos alteraciones conotruncales que más frecuentemente se diagnostican antes del nacimiento. En general son bien toleradas in utero, pero en la vida extrauterina corresponden a la principal causa de cianosis de origen cardíaco. El diagnóstico prenatal generalmente se asocia con una imagen normal del corte de cuatro cámaras, requiriendo siempre un examen detallado de los tractos de salida, idealmente utilizando Doppler color.
La tetralogía de Fallot (TDF) es la CC conotruncal más frecuente, siendo la primera causa de cianosis en la infancia por CC. Tiene una prevalencia de 1 de cada 3600 recién nacidos vivos; y representa el 3 al 7% de los niños con CC. Se caracteriza por una comunicación interventricular (CIV) subaórtica desalineada, el cabalgamiento de la raíz de la aórtica sobre la CIV y una estenosis pulmonar infundibular. La hipertrofia del ventrículo derecho, la cuarta característica anatómica de la tetralogía, típicamente no esta presente en el período prenatal. La forma típica es la TDF con estenosis pulmonar, pero el espectro de esta enfermedad incluye formas graves tales como la TDF con atresia pulmonar y la TDF con ausencia de la válvula pulmonar.
La forma clásica compuesta por estenosis pulmonar se observó en el 80% de todos lo recién nacidos con TDF. Con respecto a los hallazgos ecográficos, la vista de cuatro cámaras se aprecia normal a menos que la CIV sea grande y visible en este plano. En alrededor de un tercio de los casos es posible identificar una discreta desviación del eje cardiaco hacia la izquierda. La TDF se detecta típicamente en el corte de cinco cámaras que muestra una CIV subaórtica perimembranosa con cabalgamiento de la raíz aórtica. Este cabalgamiento se debe a una discontinuidad entre el tabique interventricular y la pared aórtica medial (también denominada CIV desalineada), con una conexión parcial de la aorta con el ventrículo derecho. De esta manera, la aorta esta desplazada hacia la derecha, y por lo general su raíz se muestra dilatada ya que recibe sangre tanto del ventrículo derecho como del izquierdo, lo cual puede brindar el primer indicio de la presencia de TDF. Además, se observa cómo el cabalgamiento de la aorta adopta un recorrido paralelo al tabique interventricular, a diferencia de la aorta ascendente en un corazón normal (Figura 1, 2 y 3). El diagnóstico de TDF también requiere la demostración de una arteria pulmonar principal estrecha pero permeable (estenosis pulmonar) que se percibe mejor en la vista del eje corto o de tres vasos. En algunas formas leves de TDF, en especial en el segundo trimestre de gestación, la discrepancia de tamaño entre el tronco pulmonar y la aorta puede ser sutil, pasándose por alto en la ecografía durante este período. El estudio con Doppler color permite demostrar la CIV (shunt de sangre a través de la CIV) y confirma la presencia del cabalgamiento de la aorta con flujo de sangre desde ambos ventrículos hacia su raíz (signo de la Y). En la vista de tres vasos se puede visualizar una arteria pulmonar de pequeño tamaño. El flujo a través del conducto arterioso es anterógrado en la TDF leve pero también puede estar invertido en los casos más graves. En tales situaciones la circulación pulmonar posnatal depende del conducto arterioso y esto se puede asociar a cianosis en el recién nacido.
En esta patología son frecuentes las anomalías cardíacas y extracardíacas asociadas. En el 83% de los casos se encuentra un foramen oval permeable o una comunicación interauricular. La asociación con arco aórtico derecho y la presencia de vena cava superior izquierda persistente ocurre en el 25 % y el 11% de los casos de TDF respectivamente.
La tasa de anomalías cromosómicas es de alrededor del 30%. La microdeleción 22q11 se encuentra en el 10-15 % de los fetos con TDF. Los signos de mal pronóstico incluyen el crecimiento lento de la arteria pulmonar, el aumento de tamaño acelerado de la aorta ascendente, la detención del flujo anterógrado a través de la válvula pulmonar y la inversión del flujo a través del conducto arterioso. En términos generales, la tetralogía de Fallot es una enfermedad que tiene buen pronóstico, y que requiere de una cirugía reparadora con sobrevida a corto y a largo plazo superiores al 90%. En los casos complejos, asociados a atresia de la válvula pulmonar el pronóstico es reservado.






")
")